



Imagen de microscopía que muestra el aspecto de un organoide cerebral derivado de células humanas. Las células en verde son progenitoras de neuronas y las células en rojo son las neuronas (imagen: Fábio Papes/Unicamp)
Investigadores de la Universidad de Campinas y de la Universidad de California develaron el mecanismo que causa el síndrome de Pitt-Hopkins, que comparte características con los trastornos del espectro autista. Los científicos revirtieron la evolución del cuadro en laboratorio, lo que allana el camino hacia el desarrollo de una terapia génica y farmacológica
Investigadores de la Universidad de Campinas y de la Universidad de California develaron el mecanismo que causa el síndrome de Pitt-Hopkins, que comparte características con los trastornos del espectro autista. Los científicos revirtieron la evolución del cuadro en laboratorio, lo que allana el camino hacia el desarrollo de una terapia génica y farmacológica
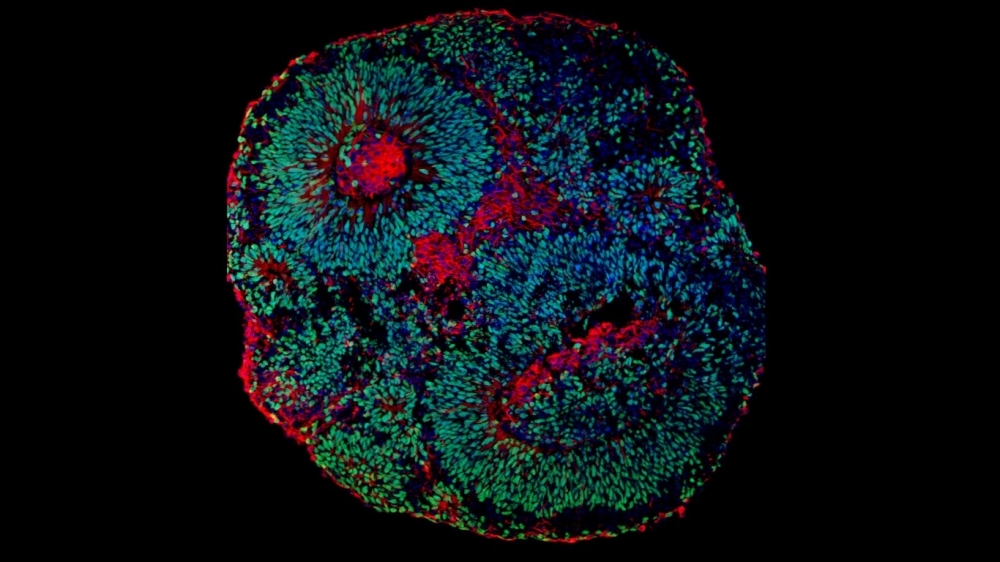
Imagen de microscopía que muestra el aspecto de un organoide cerebral derivado de células humanas. Las células en verde son progenitoras de neuronas y las células en rojo son las neuronas (imagen: Fábio Papes/Unicamp)
Por André Julião | Agência FAPESP – Un grupo de científicos encabezado por investigadores de la Universidad de Campinas (Unicamp), en el estado de São Paulo, Brasil, y de la Universidad de California en San Diego, Estados Unidos, develó el mecanismo causante del síndrome de Pitt-Hopkins, una disfunción neuropsiquiátrica con características de trastorno del espectro autista (TEA). Asimismo, los investigadores lograron revertir la evolución de dicho síndrome en modelos de laboratorio, lo que abre nuevas posibilidades de tratamiento.
Este trabajo, que cuenta con el apoyo por la FAPESP, salió publicado en la revista Nature Communications.
“Para la mayoría de los casos de TEA, no se sabe qué gen causa esa condición cuando muta. Y así lo es también en la mayoría de las enfermedades neuropsiquiátricas, tales como la esquizofrenia, la depresión y el trastorno bipolar. El síndrome de Pitt-Hopkins, a su vez, tiene su origen en una mutación en el gen TCF4. Pero hasta ahora no se conocían sus mecanismos moleculares, es decir, qué es lo que aparece diferente en las células del sistema nervioso de los pacientes con esta mutación”, comenta Fabio Papes, docente del Instituto de Biología (IB-Unicamp) y uno de los coordinadores del estudio.
Así y todo, el grupo encabezado por Papes y por Alysson Muotri, docente de la Universidad de California en San Diego, fue más allá del descubrimiento del mecanismo causante de esta condición.
Los científicos pusieron a prueba maneras de interferir en la evolución del cuadro y lograron revertir los efectos causados por la mutación. El éxito obtenido en los experimentos allana el camino hacia el desarrollo tanto de medicamentos como de una terapia génica.
El síndrome de Pitt-Hopkins se caracteriza por un déficit cognitivo, un retraso motor profundo, ausencia de habla funcional y anormalidades respiratorias, entre otras cosas. Se lo describió en el año 1978, pero su gen causante se conoció solo en 2007. Se estima que esta mutación en el gen TCF4 aparece en uno cada 35 mil nacimientos.
Minicerebros
Dado que este síndrome no se desarrolla en los ratones de la misma manera que en los humanos, el estudio en animales es inviable. Por eso los investigadores utilizaron los llamados organoides cerebrales, agrupamientos de células humanas que crecen en laboratorio y que se asemeja a miniaturas de cerebros en desarrollo, pero sin vascularización y con menos tipos celulares.
“Los organoides cerebrales constituyen un modelo más representativo que cualquier otro para estudiar disfunciones del sistema nervioso central. En este caso, las células obtenidas son de los propios pacientes. Además, los organoides son tridimensionales, por ende, su funcionamiento se ubica más cerca de la realidad que el de las células cultivadas en placas, que crecen solamente en dos dimensiones”, explica Papes.
Los organoides se generaron a partir de biopsias de la piel de portadores del síndrome, obtenidas con pacientes reclutados en la Unicamp y en Estados Unidos, aparte de sus padres, que hicieron las veces de grupo de control.
Las células se cultivaron para extraerles los llamados fibroblastos, que se transforman en células madre pluripotentes, las cuales, a su vez, pueden generar diversos tipos de células humanas. En este caso, dieron origen a neuronas, células progenitoras del sistema nervioso central y organoides cerebrales.
Mientras que las células de los padres de los pacientes formaron organoides que se desarrollaban normalmente, las de los portadores del síndrome crecían menos, como resultado de la menor replicación de las células causada por la mutación y de un detrimento de la propia neurogénesis. En otras palabras, la generación de neuronas se vio perjudicada a causa de la mutación.
Asimismo, las neuronas de los organoides con la mutación en el TCF4 aparecían en menor cantidad y exhibían menor actividad eléctrica en comparación con las de los organoides de control. Se sabe que la comunicación entre esas células se concreta con base en impulsos eléctricos, sin los cuales las mismas no pueden ejercer sus funciones. Por ende, este hallazgo puede explicar muchas características clínicas de los pacientes.
Los resultados fueron análogos a los obtenidos en tejidos del cerebro de un paciente portador de esta condición que falleció por otras razones, lo que refuerza las conclusiones obtenidas con los organoides. Este estudio fue el primero del cual se tenga noticias en el que se estudia el cerebro de una persona con síndrome de Pitt-Hopkins.
“El acceso al cerebro post mortem resultó esencial para validar algunos de los resultados obtenidos con los organoides cerebrales. El hecho de haber visto características similares entre el organoide creado en laboratorio y el cerebro muestra cuán relevante es esta tecnología”, afirma Muotri.
Una terapia génica
Una vez observadas las alteraciones causadas por la mutación en el gen TCF4, los investigadores buscaron maneras de corregirla, a los efectos de realizar lo que denominan una prueba de concepto de lo que sería un tratamiento.
Se pusieron a prueba tres intervenciones, una de ellas utilizando la técnica de edición génica conocida como CRISPR/Cas9, cuyas creadoras ganaron el Premio Nobel de Química en 2020.
Para la estrategia con CRISPR, se empleó una versión reciente de la técnica a los efectos de hacer que la copia funcional del gen existente en la célula disfuncional pase a expresar muchas más proteínas, compensando a la copia afectada por la mutación causante del síndrome de Pitt-Hopkins.
En otra intervención, con una técnica distinta, los científicos insertaron una copia extra del gen, que pasó a ejercer normalmente las funciones génicas, compensando a la copia mutada.
“Nuestro genoma posee dos copias de cada gen. Lo que causa el síndrome de Pitt-Hopkins es el hecho de que una de las copias del TCF4 no funciona. La inserción de una tercera copia o al hacer que la única copia funcional exprese más proteínas para compensar a la defectuosa puede solucionar el problema”, dice el investigador.
Los organoides que sufrieron las intervenciones empezaron a crecer normalmente y exhibieron un incremento de la proliferación de las células progenitoras, que en el cerebro dan origen a distintos tipos de células, neuronas inclusive.
“Si bien se considera que este trastorno es raro, existen otros que comprenden mutaciones en ese mismo gen. Por ende, lo que hemos descubierto podrá aplicarse en el futuro a trastornos como la esquizofrenia, por ejemplo”, afirma Papes.
Una tercera intervención consistió en la aplicación de una droga utilizada en estudios con células tumorales. Conocida con las siglas CHIR99021, la misma activa una vía de señalización celular llamada Wnt, sumamente estudiada en el contexto del cáncer. Los autores descubrieron que también se alterada debido a mutaciones en el gen TCF4.
En células y organoides disfuncionales tratados con esta droga se registró una mejoría en algunos indicadores moleculares y un aumento de tamaño (en el caso de los organoides). Estos resultados allanan el camino hacia el desarrollo de medicamentos similares que puedan tratar esta disfunción, toda vez que la CHIR99021 aún no puede emplearse en seres humanos.
“Esta vía tratada con la droga es solamente una de las que altera la mutación en el gen TCF4. La ventaja de una terapia génica con relación a un tratamiento farmacológico reside en que resolvería el problema en su origen. Así y todo, la búsqueda de nuevas drogas también es prometedora”, dice Papes.
La investigación ahora debe avanzar hacia los estudios preclínicos y clínicos. Los científicos sellaron una asociación con una empresa especializada en terapia génica, que está licenciando la tecnología empleada en los experimentos para que futuramente pueda testeársela en humanos.
Este trabajo contó también con el apoyo de la FAPESP a través de una beca de maestría otorgada a José Ricardo Teixeira Júnior y una de doctorado concedida a Antônio Camargo, ambos estudiantes de posgrado del IB-Unicamp.
La investigación tuvo también el apoyo del Consejo Nacional de Desarrollo Científico y Tecnológico (CNPq) de Brasil, de los National Institutes of Health (NIH) de Estados Unidos y de la Pitt-Hopkins Research Foundation.
Puede leerse el artículo intitulado Transcription Factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content en el siguiente enlace: www.nature.com/articles/s41467-022-29942-w.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
