



Trabalho coordenado por pesquisador da Unesp facilita a análise de materiais constituídos por um grande número de átomos estruturados tridimensionalmente; um exemplo é o molibdato de bário, que tem potencial aplicação tecnológica em luminescência e em degradação de compostos orgânicos (imagem: Ricardo Luis Tranquilin / Unesp)
Trabalho coordenado por pesquisador da Unesp facilita a análise de materiais constituídos por um grande número de átomos estruturados tridimensionalmente; um exemplo é o molibdato de bário, que tem potencial aplicação tecnológica em luminescência e em degradação de compostos orgânicos
Trabalho coordenado por pesquisador da Unesp facilita a análise de materiais constituídos por um grande número de átomos estruturados tridimensionalmente; um exemplo é o molibdato de bário, que tem potencial aplicação tecnológica em luminescência e em degradação de compostos orgânicos
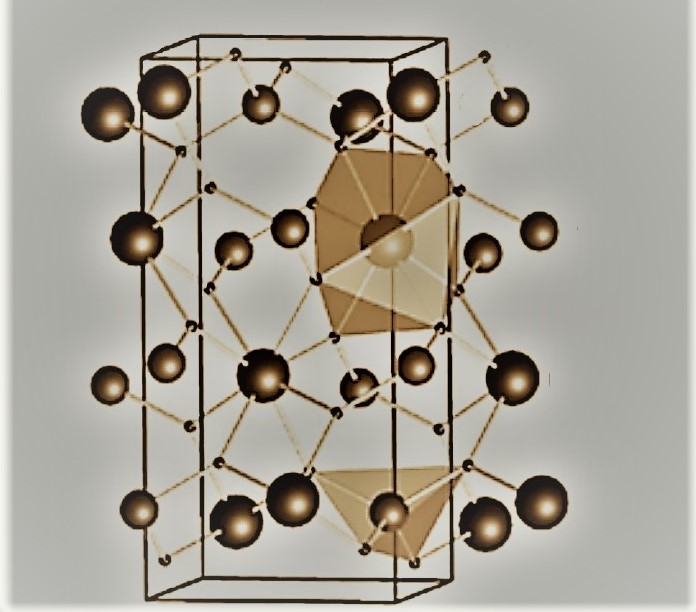
Trabalho coordenado por pesquisador da Unesp facilita a análise de materiais constituídos por um grande número de átomos estruturados tridimensionalmente; um exemplo é o molibdato de bário, que tem potencial aplicação tecnológica em luminescência e em degradação de compostos orgânicos (imagem: Ricardo Luis Tranquilin / Unesp)
José Tadeu Arantes | Agência FAPESP – Descrever e explicar as propriedades eletrônicas de certos materiais cristalinos, constituídos de estrutura atômica ordenada tridimensionalmente, pode ser bastante complicado em razão da grande quantidade de átomos envolvidos, cada um deles com um grande número de elétrons. Modelos simplificados têm sido desenvolvidos com essa finalidade. Foi o que aconteceu com a Teoria do Funcional de Densidade (DFT, do inglês Density Functional Theory), derivada da mecânica quântica e usada em física dos sólidos e em química para resolver sistemas de muitos corpos. Na DFT, as propriedades de sistemas com muitos elétrons são determinadas por meio de funcionais, isto é, de funções de outra função, que, no caso, é a distribuição espacial da densidade eletrônica.
Proposta em um artigo publicado em 1964 pelo físico austro-americano Walter Kohn (1923-2016) e pelo físico franco-americano Pierre Hohenberg (1934-2017), a Teoria do Funcional de Densidade tornou-se amplamente aplicada graças ao desenvolvimento dos recursos computacionais. Em 1998, Kohn recebeu o Prêmio Nobel de Química em reconhecimento por seu papel na criação da DFT.
Mas ainda é necessário aprimorar os métodos para obter uma modelagem computacional mais realista. Este foi o objetivo de um estudo apoiado pela FAPESP e publicado no periódico Computational Materials Science e liderado por Julio Ricardo Sambrano, coordenador do grupo de Modelagem e Simulação Computacional da Faculdade de Ciências da Universidade Estadual Paulista (Unesp) em Bauru. O trabalho teve a colaboração do professor Elson Longo, diretor do Centro de Desenvolvimento de Materiais Funcionais (CDMF), um Centro de Pesquisa, Inovação e Difusão (CEPID) apoiado pela FAPESP na Universidade Federal de São Carlos (UFSCar).
O material considerado no estudo foi o molibidato de bário (BaMoO4), escolhido por sua potencial aplicação tecnológica em luminescência e em degradação de compostos orgânicos. Atualmente, o BaMoO4 é usado para melhorar a adesão de esmaltes e para remover o enxofre da nafta (derivado do petróleo) na produção de gases.
A Agência FAPESP conversou com os professores Sambrano e Longo sobre o trabalho agora publicado.
Agência FAPESP – Por que é difícil descrever e explicar as propriedades de certos materiais cristalinos?
Julio Ricardo Sambrano – Os materiais cristalinos têm uma estrutura ordenada tridimensionalmente, com uma enormidade de átomos, cada um dos quais constituídos por vários elétrons. E cada um desses elétrons pode ser representado por uma função matemática. A situação ideal é a obtenção de um modelo computacional que consiga descrever fielmente o sistema cristalino, isto é, conceber um modelo que considera todas as forças atuantes no sistema. Infelizmente, as limitações atuais dos modelos teóricos e a capacidade computacional disponível fazem com que essa tarefa seja impossível de ser completada. Possivelmente, em um futuro próximo e com o desenvolvimento da computação quântica, isso possa ser modificado. Como exemplo, atualmente, os cálculos mecânico-quânticos fazem uma série de aproximações – a começar, considerando os núcleos atômicos como fixos, quando, na prática, eles estão em constante movimento. As interações entre os elétrons também são aproximadas para dois elétrons.
Agência FAPESP – Como a Teoria do Funcional da Densidade (DFT) ajuda a contornar o problema?
Elson Longo – No início da mecânica quântica, poucos sistemas podiam ser resolvidos analiticamente ou mesmo por meio de uma solução numérica aproximada. No entanto, em 1927, o físico britânico Llewellyn Thomas (1903-1992) e o físico italiano Enrico Fermi (1901-1954) propuseram um modelo probabilístico para análise da densidade eletrônica dos átomos. Dessa ideia, surgiu, décadas mais tarde, a Teoria do Funcional de Densidade, elaborada por Kohn e Hohenberg. Esse modelo abriu duas portas: a viabilização dos cálculos de sistemas complexos como os cristais e a facilitação da interpretação dos resultados. Assim, de forma coerente, pode-se interpretar as propriedades de sistemas tridimensionais, de superfícies bidimensionais ou mesmo sistemas unidimensionais, como nanotubos e nanofios, e compará-las com os resultados experimentais.
Agência FAPESP – Por que a DFT é uma alternativa limitada?
Longo – Não somente a DFT é uma alternativa que ainda tem limitações, como toda a mecânica quântica está em constante avanço em função do desenvolvimento de novas alternativas e metodologias. Isso devido à maior disponibilidade computacional, que, no caso, possibilita o cálculo de sistemas cristalinos mais complexos; o desenvolvimento de algoritmos mais sofisticados; e a uma maior correlação entre os modelos e os resultados experimentais.
Agência FAPESP – Que novos métodos podem proporcionar uma representação mais acurada dos materiais de interesse?
Sambrano – O avanço atual da mecânica quântica está sendo pautado pela interpretação dos dados teóricos em função dos resultados experimentais. O conhecimento das energias das diferentes superfícies de um cristal constitui uma ferramenta importante para produzir modelos teóricos das morfologias de um cristal em escala nanométrica. Essas morfologias podem ser associadas aos resultados experimentais obtidos em microscopia eletrônica de alta resolução. E os resultados podem ser associados às propriedades dos cristais.
Agência FAPESP – Quais são as possíveis aplicações tecnológicas de uma representação mais acurada?
Longo – Para que haja geração de tecnologia, o primeiro passo é o desenvolvimento do conhecimento. O refinamento dos modelos teóricos possibilita melhor compreensão de como uma propriedade sofre transformação, em função da perturbação de sua densidade eletrônica. Dessa forma, pode-se conhecer melhor um cristal e propor novas pesquisas direcionadas a uma propriedade específica. Por exemplo, melhorar o poder bactericida de um cristal e, simultaneamente, proporcionar uma melhor resposta de suas propriedades fotoluminescentes.
Agência FAPESP – Qual foi o programa computacional usado no estudo?
Longo – O programa foi o CRYSTAL17, desenvolvido pelo grupo coordenado pelo professor Roberto Dovesi na Università degli Studi di Torino, em Turim, na Itália. Esse programa permite executar simulações em nível DFT de cálculos mecânicos quânticos para estruturas periódicas, tais como a dos cristais, e obter as propriedades relacionadas para esses sistemas. O grupo de pesquisa coordenado pelo professor Sambrano na Unesp tem ampla experiência na área de simulações aplicadas a materiais e no programa CRYSTAL. O professor Sambrano passou um longo período em Turim com o professor Dovesi e mantém colaboração com os desenvolvedores do programa. Essa parceria já rendeu uma série de artigos publicados, cujos temas estão relacionados com as pesquisas do CDMF apoiadas pela FAPESP.
O artigo Computational procedure to an accurate DFT simulation to solid state systems pode ser acessado em https://www.sciencedirect.com/science/article/abs/pii/S0927025619304756?via%3Dihub#!.
A Agência FAPESP licencia notícias via Creative Commons (CC-BY-NC-ND) para que possam ser republicadas gratuitamente e de forma simples por outros veículos digitais ou impressos. A Agência FAPESP deve ser creditada como a fonte do conteúdo que está sendo republicado e o nome do repórter (quando houver) deve ser atribuído. O uso do botão HMTL abaixo permite o atendimento a essas normas, detalhadas na Política de Republicação Digital FAPESP.
