



Research led by a Brazilian scientist facilitates the analysis of materials comprising a large number of three-dimensionally structured atoms, such as barium molybdate, with potential applications in luminescence and organic compound degradation (image: Ricardo Luis Tranquilin / UNESP)
Research led by a Brazilian scientist facilitates the analysis of materials comprising a large number of three-dimensionally structured atoms, such as barium molybdate, with potential applications in luminescence and organic compound degradation.
Research led by a Brazilian scientist facilitates the analysis of materials comprising a large number of three-dimensionally structured atoms, such as barium molybdate, with potential applications in luminescence and organic compound degradation.
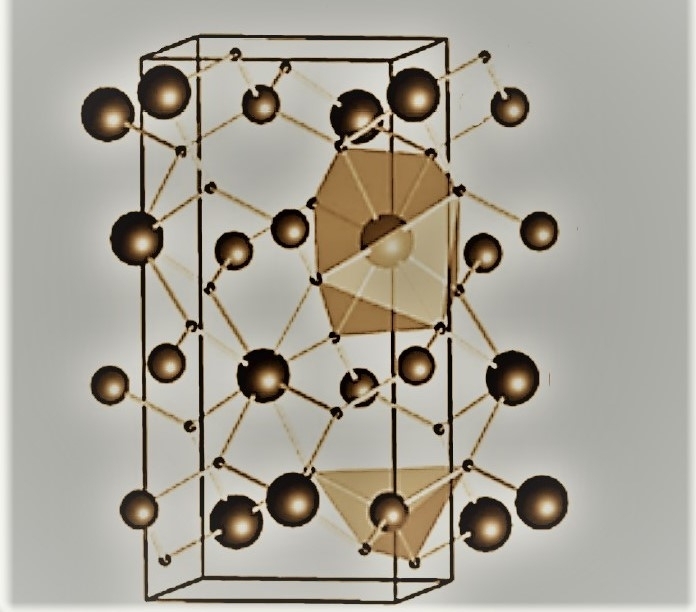
Research led by a Brazilian scientist facilitates the analysis of materials comprising a large number of three-dimensionally structured atoms, such as barium molybdate, with potential applications in luminescence and organic compound degradation (image: Ricardo Luis Tranquilin / UNESP)
By José Tadeu Arantes | Agência FAPESP – Describing and explaining the electronic properties of certain crystalline materials that have a three-dimensionally ordered atomic structure can be complicated owing to the large number of atoms involved, each with a large number of electrons. Simplified models that have been developed for this purpose include density functional theory (DFT), derived from quantum mechanics and used in solid-state physics and chemistry to resolve many-body systems. In DFT, the properties of systems with many electrons are determined by means of functionals, i.e., functions of another function, which in this case is the spatial distribution of the electron density.
First proposed in an article published in 1964 by Austrian-born American physicist and chemist Walter Kohn (1923-2016) and French-born American physicist Pierre Hohenberg (1934-2017), DFT has been widely applied thanks to advances in computational resources. Kohn was awarded the 1998 Nobel Prize for Chemistry in recognition of his role in creating DFT.
However, the methods still need to be refined to obtain more realistic computational modeling. This was the goal of a study conducted in Brazil with support by FAPESP, whose results were published in Computational Materials Science. The principal investigator for the study was Julio Ricardo Sambrano, who leads the Computational Modeling and Simulation Group at São Paulo State University’s School of Sciences (FC-UNESP) at Bauru. Another scientist who made an important contribution to the study was Professor Elson Longo, director of the Center for the Development of Functional Materials (CDMF), one of the Research, Innovation and Dissemination Centers (RIDCs) supported by FAPESP. The CDMF is hosted by the Federal University of São Carlos (UFSCar) in São Paulo State, Brazil.
The material considered in the study was barium molybdate (BaMoO4), chosen for its potential technological application to luminescence and to the degradation of organic compounds. BaMoO4 is currently used as an adhesive agent for enamelware and to remove sulfur from naphtha (a refined petroleum product) in the production of gases.
Agência FAPESP interviewed Sambrano and Longo about the study.
Agência FAPESP – Why is it difficult to describe and explain the properties of certain crystalline materials?
Julio Ricardo Sambrano – Crystalline materials have a three-dimensionally ordered structure with a large number of atoms, each of which consists of several electrons, and each of these electrons can be represented by a mathematical function. Ideally, we should be able to obtain a computational model that accurately describes the crystal system, i.e., to design a model that considers all the forces at work in the system. Unfortunately, however, this task just is not feasible with the theoretical models we have and the computing capacity available. In the near future, it may be feasible, especially when quantum computing is developed further. For example, quantum mechanics calculations currently perform certain approximations, such as considering atomic nuclei to be fixed, whereas in fact they are constantly moving. Interactions among electrons are also approximated to two electrons.
Agência FAPESP – How does density functional theory (DFT) help surmount this problem?
Elson Longo – In the early days of quantum mechanics, few systems could be resolved analytically or even by means of a numerically approximate solution. However, in 1927, British physicist Llewellyn Thomas (1903-1992) and Italian physicist Enrico Fermi (1901-1954) proposed a probabilistic model for the analysis of the electron density of atoms. Decades later, this idea led to DFT, developed by Kohn and Hohenberg. It enables us to compute complex systems such as crystals and facilitates interpretation of the results. Therefore, we can coherently interpret the properties of three-dimensional systems, two-dimensional surfaces, and even one-dimensional systems such as nanotubes and nanowires and compare them with experimental results.
Agência FAPESP – Why is DFT a limited alternative?
Longo – Not only is DFT an alternative that still has limitations, but also quantum mechanics in its entirety is constantly advancing thanks to the development of new alternatives and methodologies. This is made possible by the expansion of computer power, enabling, in this case, the calculation of more complex crystal systems, the development of more sophisticated algorithms, and the obtaining of a closer correlation between models and experimental results.
Agência FAPESP – What new methods can obtain a more accurate representation of the materials of interest?
Sambrano – Current advances in quantum mechanics are being driven by the better interpretation of theoretical data on the basis of experimental results. Knowledge of the energies of the various surfaces of a crystal is important to produce theoretical models of the morphologies of a crystal on the nanometric scale. These morphologies can be associated with the experimental results obtained using high-resolution electron microscopy, and the results can be associated with the properties of crystals.
Agência FAPESP – What are the possible technological applications of a more accurate representation?
Longo – The first step in developing any technology is knowledge creation. Here, by refining the theoretical models, we are able to achieve a better understanding of how properties change when the electron density is disturbed. Our knowledge of crystal systems increases, and we can propose new research to target a specific property, for example, improving the bactericidal power of a crystal and, at the same time, producing a better response to its photoluminescent properties.
Agência FAPESP – Tell us about the computer program used in the study.
Longo – The program was CRYSTAL17, developed by Professor Roberto Dovesi and his group at the University of Turin in Italy. This program runs DFT simulations of quantum mechanics calculations for periodic structures such as those of crystals and computes the related properties for these systems. Sambrano’s research group at UNESP has ample experience with simulations applied to materials and with the program. Sambrano spent a long period in Turin with Dovesi and continues to work with the developers of the program. This partnership has already led to several publications on topics linked to CDMF’s research projects supported by FAPESP.
The article “Computational procedure to an accurate DFT simulation to solid state systems” can be retrieved from: www.sciencedirect.com/science/article/abs/pii/S0927025619304756?via%3Dihub#!.
The Agency FAPESP licenses news via Creative Commons (CC-BY-NC-ND) so that they can be republished free of charge and in a simple way by other digital or printed vehicles. Agência FAPESP must be credited as the source of the content being republished and the name of the reporter (if any) must be attributed. Using the HMTL button below allows compliance with these rules, detailed in Digital Republishing Policy FAPESP.
